
Behandling
Polycytemia vera (PV)
Rekommendationer
- Alla patienter bör behandlas med:
- Flebotomi med mål EVF <45 % (++++)
- Lågdos ASA 75 mg/dag (++++)
- Cytoreduktiv behandling bör ges vid:
- Högrisksjukdom, dvs. ålder >60 år och/eller tidigare trombotisk händelse (+++)
- Nedsatt tolerans för bibehållande av EVF <45 % med enbart flebotomi (++)
- PV-relaterade symtom inklusive symtomatisk mjältförstoring (++)
- Proliferativ sjukdom med 100 % respektive 50 % ökning LPK (vid utgångsvärde <10 resp >10), LPK >15 36 och/eller TPK >1 500 (++)
- Rekommenderade förstahandsval vid cytoreduktiv behandling:
- Hydroxyurea (+++) eller IFN-alfa (++) (se resonemang i text)
- Vid hög ålder eller kort förväntad överlevnad hydroxyurea (+++) eller busulfan (++)
- Riskfaktorer för hjärt-kärlsjukdom bör behandlas (++)
Mål med behandling
Behandling av PV syftar till att minska PV-relaterade symtom och minska risken för hjärt-kärlhändelser. Vid behandling av yngre patienter är även minskning av risken för transformation och förlängd överlevnad viktiga mål. Behandlingen styrs i hög grad av riskvärderingen.
Utöver sjukdomsspecifik behandling bör övriga riskfaktorer för hjärt-kärlsjukdom beaktas, och då särskilt rökning och hypertoni som är överrepresenterat hos PV-patienter 37 och utgör en oberoende riskfaktor för hjärt-kärlhändelser 38.
Vid cytoreduktiv behandling eftersträvas normalisering av LPK och TPK 39. Rekommendationen innebär dock att cytoreduktiv behandling i vissa fall bör ges till patienter med normala blodvärden. I dessa fall bör behandlingen anpassas så att anemi, blödnings- och infektionsproblem undviks genom att man anpassar doseringen så att blodvärdena ligger i det nedre normalintervallet.
Riskstratifiering och indikation för behandling
Riskstratifiering vid PV utgår ifrån risken att drabbas av en hjärt-kärlhändelse och leder till indelning i de två kategorierna lågrisk- och högrisksjukdom.
Högrisksjukdom föreligger vid:
- Ålder >60 år och/eller
- Tidigare hjärt-kärlhändelse 2.
Lågrisksjukdom föreligger vid:
- Ålder <60 år och
- Frånvaro av tidigare hjärt-kärlhändelse 40.
Utöver riskgrupp är leukocytos 41 samt de traditionella riskfaktorerna för hjärt-kärlsjukdom, riskfaktorer för att drabbas av hjärt-kärlhändelser vid PV.
Nyare risk-scores för överlevnad, vilka även inkluderar molekylära markörer (MIPPS-PV-score) har presenterats men används för nuvarande ej i klinisk praxis 42.
Alla PV-patienter bör behandlas med flebotomi och lågdos acetylsalicylsyra (ASA). Generellt bör cytoreduktiv behandling ges vid högrisksjukdom. Vid lågrisksjukdom rekommenderas behandling vid nedsatt tolerans för flebotomi som medför svårigheter att bibehålla EVF-mål (6 eller fler venesektio per år), PV-relaterade symtom inklusive symtomatisk mjältförstoring, proliferativ sjukdomsbild med progessiv leukocytos (100 % ökning vid utgångsvärde <10 eller minst 50 % ökning vid utgångsvärde > 10), ihållande leukocytos med LPK > 15 och/eller TPK > 1 500 36.
Flebotomi
I en randomiserad klinisk prövning 43 inkluderande låg- och högrisk PV har flebotomi med målnivå för hematokrit på EVF <45 % jämförts med målnivå EVF 45–50 %. Studien visar minskad dödlighet i hjärt-kärlsjukdom med det lägre EVF-målet.
Vid höga EVF och/eller Hb-nivåer >0,55 respektive >190 som inte har någon uppenbar förklaring som exempelvis intorkning, bör skyndsamma åtgärder övervägas. Flebotomi med 400 ml blod kan göras. 500 ml dryck per os eller vid behov som intravenös infusion ges efter tappning. Observera att EPO-provtagning bör tas innan eventuell flebotomi.
Acetylsalicylsyra
Acetylsalicylsyra (ASA) är ett effektivt läkemedel mot mikrovaskulära symtom. Vidare har lågdos ASA i en randomiserad studie 36 vid PV utan annan indikation för ASA, visat ett minskat antal trombotiska händelser utan att signifikant öka risken för blödning.
Man bör vara försiktig vid behandling av patienter med ett trombocytvärde över 1 000 på grund av ökad blödningsrisk 44. För patienter med samtidig indikation för antikoagulantiabehandling rekommenderas i normalfallet enbart antikoagulantiabehandling 4546 (++). För patienter med intolerans för ASA rekommenderas klopidogrel (+).
Hydroxyurea
Hydroxyurea (HU) är ett cytoreduktivt läkemedel som kan förebygga hjärt-kärlhändelser vid PV 22474849 och har dokumenterade långtidsdata 50. HU är ett milt cytostatikum verkande på DNA replikationen. Utifrån verkningsmekanismen finns en teoretisk risk att läkemedlet skulle kunna öka risken för transformation till akut leukemi, dock har prospektiva studier enbart kunnat visa en ökad leukemiförekomst hos patienter vilka tidigare behandlats med andra cytostatika 51. Vidare har en svensk registerstudie inte kunnat påvisa ökad leukemirisk efter HU behandling 52.
Vid behandling av yngre patienter rekommenderas att interferonbehandling övervägas i första hand. HU ska inte användas under graviditet. Behandling inleds med 500–1000 mg/d. Läkemedlet tolereras i allmänhet väl. De biverkningar man bör vara observant på under HU behandling är bensår, feber och kraftigt svängande trombocytvärden.
Interferon
Pegylerat interferon-alfa (IFN-alfa) har en lång behandlingstradition i Sverige. Det tillgängliga läkemedlet peginterferon alfa-2a har dock enbart indikation för behandling av hepatit C. Flera fas 2-studier har visat att IFN-alfa kan inducera långvariga hematologiska och molekylära remissioner vid PV 535455. IFN-alfa kan effektivt minska PV-relaterade symtom 56 och förefaller skydda mot hjärt-kärlhändelser vid utvärdering i en studie som kombinerat effektmått 57. Generellt är interferon den rekommenderade förstalinjebehandlingen för patienter under 60 år 2.
Ett nytt pegylerat IFN-alfa-preparat, ropeginterferon alfa-2b, har visat jämförbara resultat med hydroxyurea i en randomiserad fas 3-studie 58. Läkemedlet är godkänt i Sverige men saknar subvention varvid läkemedlet ej kan rekommenderas.
Peginterferon alfa-2a ges som subkutan injektion varje eller var annan vecka, lämplig startdos är (45) –90 mgr /v eller 135 mgr var annan vecka. Majoriteten av patienterna svarar på motsvarande en veckodos om 90–135 mgr inom 2–3 månader. Efter att ett behandlingssvar har uppnåtts kan i många fall behandlingsintensiteten minskas över tid.
Försiktighet bör iakttas vid autoimmun eller psykiatrisk sjukdom. Lever- och tyreoideavärden bör följas under interferonbehandling. Vanliga biverkningar är influensaliknande besvär. Om hypothyreos skulle utvecklas under behandlingen kan INF behandling i vissa fall fortsätta med tillägg av Levaxin förutsatt att patienten har haft god effekt av INF och att övriga biverkningar är milda. Behandlingsavbrott bör övervägas vid tillkomst av depressiva besvär.
Busulfan
Busulfan givet intermittent i lågdos under cirka 4–6 veckor är en effektiv cytoreduktiv andra linjens behandling vid PV 59, men bör på grund av den ökade risken för leukemisk transformation endast ordineras äldre patienter.
Behandling ges med (2) – 4 mg dagligen.
Under behandlingen följs blodvärden varje vecka, och behandlingen sätts ut så fort TPK- och/eller LPK-värdena normaliseras.
Biverkningarna är vanligen milda.
Ruxolitinib
Ruxolitinib har i en randomiserad studie visat hematologisk, symtomlindrande och mjältminskande effekt hos patienter refraktära mot hydroxyurea 60. Studien var inte designad för att visa förebyggande effekt mot hjärt-kärlhändelser, men subgruppsanalyser och poolade data från andra studier talar för att sådan effekt kan finnas 61. Nyare data visar förutom bättre komplett hematologisk respons och EFS även bättre molekylär respons av Ruxolitinib jämfört med BAT och ett samband mellan molekylär respons och EFS, PFS och OS 62.
Ruxolitinib saknar dock subvention vid PV och kan därför inte rekommenderas som första linjens behandling, men kan övervägas i speciella situationer såsom vid intolerans mot rekommenderade läkemedel eller svår klåda (se Kapitel 11 Speciella situationer).
Skifte av behandling
Vid otillräcklig effekt (resistens) mot primärbehandlingen eller vid intolerans är en byte av terapi indicerat. När det gäller hydroxyurea rekommenderas ett byte av terapi om otillräckligt svar föreligger vid en lägsta dos av 1,5 – 2 g/dag under minst 4 månader 63.
Ett byte av den cytoreduktiva behandlingen vid otillräcklig effekt bör övervägas vid:
- Ökning av leukocytantalet (minst 100 % ökning av leukocytantalet vid utgångsvärde <10 och minst 50 % ökning vid utgångsvärde >10).
- Ihållande leukocytos (>15, bekräftat efter 3 månader).
- TPK >1 000.
- Mikrocirkulationsstörningar under längre tid än 3 månader.
- Otillräcklig kontroll av hematokrit under underhållsfasen med venesectio för att hålla hematokriten under 45 % (6 eller fler venesectio per år).
- Symtomatisk eller progressiv splenomegali med ökning av mjältstorlek med 5 cm på 1 år.
- Allvarliga eller påfrestande symtom (till exempel ihållande klåda).
- Uppkomst av tromboser eller blödningar.
Prevention vid hjärt-kärlsjukdom
Hos patienter med PV bör riskfaktorer för hjärt-kärlsjukdom såsom diabetes, hypertoni och hyperlipidemi vara välbehandlade samt följas upp regelbundet 64. Rökstopp bör rekommenderas. Övriga levnadsvanor såsom hälsosam kost och regelbunden fysisk aktivitet är också viktigt för att minska risken för hjärt-kärlsjukdom, förbättra livskvaliteten och lindra symtom.
Essentiell trombocytemi (ET)
Rekommendationer
- Alla patienter bör riskkategoriseras enligt r-IPSET thrombosis för värdering av trombosrisk, JAK2 och MPL har en högre trombosrisk än CALR.
- Lågdos ASA 75 mg 1-2 gånger per dag förebygger hjärt-kärlhändelser, och bör ges till alla utom de med mycket låg risk. (++)
- Cytoreduktiv behandling
- bör övervägas till alla med intermediär risk
- bör ges till alla med hög risk enligt r-IPSET thrombosis, uttalad trombocytos (> 1 500), MPN-relaterad blödning, vid proliferativ sjukdom samt vid mikrocirkulatoriska symtom med otillräckligt svar på ASA. (+++)
- Rekommenderade förstahandsval för cytoreduktion är hydroxyurea (++++) eller interferon. (++)
- Riskfaktorer för hjärt-kärlsjukdom bör behandlas. (++)
Mål med behandling
Ett viktigt mål med behandling av ET är att minska risken för hjärt-kärlhändelser och blödning, minska ET-relaterade symtom samt hantera risksituationer såsom graviditet och kirurgi. Om cytoreduktion ges, styrs behandlingen mot normalisering av trombocytos och leukocytos, även reversering av leukocytos ter sig vara av betydelse för att minska trombosrisk 65666768.
Det finns idag inga säkra bevis på hur behandling påverkar risken för transformation till myelofibros eller akut leukemi, men en förhoppning om att interferon har en sjukdomsmodifierande effekt. Risken för transformation är liten vid WHO-definierad ET. Ett, delvis framtida, mål är ett att förhindra eller fördröja progress, men där vi ännu saknar fullständig evidens.
Ett viktigt mål med uppföljningen är att identifiera patienter med progress till myelofibros eller transformation med blastökning. Vid sjunkande perifera värden, tillkomst av leukoerytroblastos, konstitutionella symtom, växande mjälte eller annat som föranleder en misstanke om progress bör förnyad benmärgsundersökning inklusive NGS och cytogenetik göras. Fullständig genetik är särskilt viktigt hos patienter i transplantabel ålder.
Riskstratifiering och indikation för behandling
Risken för trombos bedöms utifrån r-I IPSET thrombosis, International Prognostic Score for ET, där tidigare tromboser, ålder över 60 år, JAK2-mutation utgör riskfaktorer för trombos vilket delar in patienterna i 4 riskkategorier: mycket låg, låg, intermediär och hög risk 69707172.
I figur 3 visas riskstratifiering anpassad från r-IPSET thrombosis och föreslagen behandling utifrån trombosrisk. MPL ingår inte i original-publikationen för r-IPSET thrombosis, men går med en högre trombosrisk än vad som visats hos CALR-muterade, och bör således inte betraktas som väldigt låg risk eller intermediär risk. Att avstå från ASA har bara visats vara gynnsamt hos CALR-muterade 73747576.
Trippelnegativa patienter har som grupp en lägre risk för såväl trombos som fibrotisk transformation, men är en heterogen grupp, som inkluderar individer med och utan klonalt driven hematopoes, och hos en del av dessa patienter föreligger en osäkerhet kring hurvida dessa patienter har en myeloproliferativ neoplasi. Viss försiktighet rekommenderas därför med vad gäller val av cytoreduktiva läkemedel till dessa patienter 77.
Figur 3. Riskstratifiering från r-IPSET thrombosis och föreslagen behandling utifrån trombosrisk.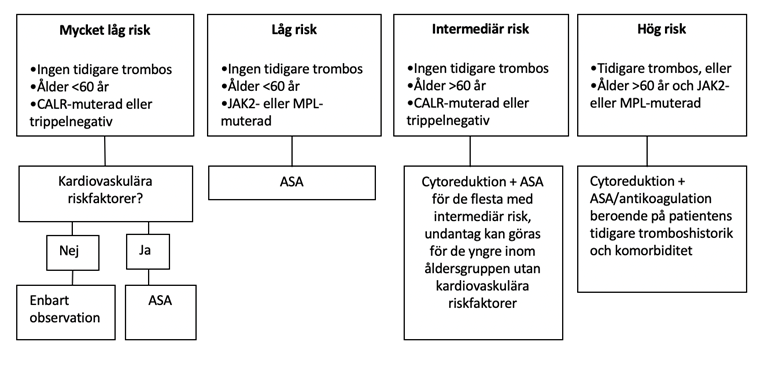
Även tidigare MPN-relaterad blödning eller ökad blödningsrisk på grund av extrem trombocytos (>cirka 1 500) utgör indikation för cytoreduktiv behandling. Proliferativ sjukdom eller mikrocirkulatoriska symtom som huvudvärk eller erytromelalgi med otillräckligt svar på ASA kan också utgöra indikation för cytoreduktiv behandling 78.
Flera risk-stratifieringsmodeller finns för överlevnad vid ET; IPSET och Triple A, inkluderandes kliniska och lab-variabler, samt MIPPS-ET som även inkluderar molekylära variabler. Gemensamt för dessa är att ålder väger tungt, och att behandling inte styrs efter dem, och de anses därför inte nödvändiga att använda kliniskt 42.
Trombocytaggregationshämmande behandling
ASA i lågdos (75 mg1-2 gånger per dag) bör ges till alla patienter med låg-, intermediär- eller högrisk utan kontraindikationer, samt till alla patienter med mikrocirkulatoriska symtom, eller med riskfaktorer för hjärt-kärlsjukdom, se figur 3 798081. Patienter med CALR-mutation utan andra riskfaktorer har dock ökad risk för blödningar och mindre nytta av ASA, och man kan hos dessa avstå från ASA 8182.
Två-dosförfarande av låg-dos ASA jämfört med en-dosförfarande har hos ET-patienter i en randomiserad studie visats ge en signifikant och påtaglig sänkning av tromboxan A2. Man såg också en signifikant minskning av mikrovaskulärt symtom, en icke-signifikant ökad trombosrisk (få events i båda grupperna) och icke-signifikant ökad blödningsrisk 83. Två-dos förfarande kan därav övervägas för patienter med en högre trombotisk/kardiovaskulär risk samt vid mikrovaskulära symtom som inte svarar på en-dos förfarande.
ASA bör användas med försiktighet vid höga trombocytvärden> 1 000 och undvikas vid trombocytvärden> 1 500 då det finns en paradoxalt ökad blödningsrisk på grund av brist på stora von Willebrand multimerer, i analogi med förvärvad von Willebrands sjukdom. Trombocytvärden> 1 500 utgör indikation för cytoreduktiv behandling 6684.
För ASA-intoleranta patienter rekommenderas klopidogrel (+).
Cytoreduktion
Om cytoreduktion är indicerat rekommenderas läkemedel enligt nedan i första hand. I randomiserade studier har hydroxyurea och pegylerat interferon visats vara likvärdigt avseende trombosrisk och komplett hematologiskt svar, histopatologiska svar är vanligare hos hydroxyureabehandlade, medan molekylära svar var vanligare i interferongrupperna 8586.
Behandlingen bör styras mot ett normaliserat blodstatus (TPK <400 och normaliserat LPK), men bör balanseras mot tolerans. Vid normalt blodstatus vid behandlingsstart bör målet vara att undvika blodvärden under gränsen för normalintervall. Patienter under 60 år utan högriskfaktorer har inte visat sig ha någon nytta av cytoreduktiv behandling 87. Ruxolitinib har ingen plats i behandlingen av ET 88.
- Hydroxyurea eller interferon-alfa i första hand.
- Interferon-alfa eller hydroxyurea i andra hand.
- Anagrelid i tredje hand om enbart behov av trombocytsänkning.
> 75 års ålder eller kort förväntad överlevnad:
- Hydroxyurea i första hand.
- Intermittent busulfan i andra hand.
Hydroxyurea
Hydroxyurea (HU) är det cytoreduktiva läkemedel som har störst evidens vid behandling av ET 89 då HU har visats kunna minska risken för hjärt-kärlhändelser 90.
HU är ett milt cytostatikum, som har visats vara en säker och tolerabel behandling och en stor svensk registerstudie har inte påvisat ökad leukemiförekomst vid HU behandling 52. Vid behandling av yngre patienter rekommenderas att inferferonbehandling övervägs i första hand. Behandling inleds med 500–1000 mg/d. Biverkningarna är milda men läkemedlet kan behöva sättas ut vid svårläkta bensår.
Interferon
Det finns en lång behandlingstradition och positiv erfarenhet av IFN-alfa vid ET 9192. IFN-alfa har inte visat någon ökad risk för leukemogenicitet eller teratogenicitet och ger en hög andel kliniska och molekylära remissioner 93. Indikation saknas i Fass för behandling av MPN, men rekommenderas enligt ovan mot bakgrund av den långvariga behandlingserfarenhet som finns. Pegylerade former bör användas på grund av glesare doseringsintervall och ökad tolerabilitet. Tillgängligt i Sverige idag finns peginterferon alfa-2a (Pegasys). En ny fas-3 studie 94 vid PV och ET visar ingen skillnad i skydd mot tromboemboliska händelser eller sjukdomsprogress mellan HU och IFN-alfa. Median uppföljningstid i denna studie var dock kort, 81 veckor.
Peginterferon alfa-2a ges som subkutan injektion varje eller var annan vecka, lämplig startdos är (45) –90 mgr /v eller 135 mgr var annan vecka. Majoriteten av patienterna svarar på motsvarande en veckodos om 90–135 mgr inom 2–3 månader. Efter att ett behandlingssvar har uppnåtts kan i många fall behandlingsintensiteten minskas över tid.
Försiktighet bör iakttas vid autoimmun eller psykiatrisk sjukdom. Lever- och tyreoideavärden bör följas under interferonbehandling. Vanliga biverkningar är influensaliknande besvär. Om hypothyreos skulle utvecklas under behandlingen kan IFN behandling i vissa fall fortsätta med tillägg av Levaxin förutsatt att patienten har haft god effekt av IFN och att övriga biverkningar är milda. Behandlingsavbrott bör övervägas vid tillkomst av depressiva besvär.
Busulfan
Busulfan är ett alkylerande cytostatikum, med god cytoreduktiv effekt och som ofta tolereras väl av äldre patienter. Det har inte helt gått att utesluta att Busulfan är associerat med en högre transformations-frekvens till akut leukemi, men sannolikt inte. Busulfan används därför företrädesvis till biologiskt äldre patienter (>cirka 75 år). Toleransen är ofta god, och det rekommenderas därför i andra hand till äldre med intolerans eller resistens mot hydroxyurea 5295.
Behandlingen ges intermittent, och startas med 2–4 mg dagligen i 4–8 veckor, tills TPK är <400 eller LPK <3, då uppehåll görs, tills nytt behandlingsbehov uppstår.
Blodstatus behöver följas tätt under behandlingen och varje till varannan månad efter avslutad behandling.
Anagrelid
Anagrelid är ett icke-cytostatiskt behandlingsalternativ med enbart trombocytsänkande effekt, utan påverkan på till exempel leukocytvärden, och bidrar till en minskad risk för hjärt-kärlhändelser 8996. Risken för blödning är ökad vid samtidig användning av ASA. Något fler fall av progress till myelofibros har noterats 8997.
Biverkningarna är framför allt kardiella och EKG bör kontrolleras inför behandlingsstart. Anagrelid bör undvikas till patienter med hjärtsjukdom.
Prevention vid hjärt-kärlsjukdom
Hos patienter med ET bör riskfaktorer för hjärt-kärlsjukdom såsom diabetes, hypertoni och hyperlipidemi vara välbehandlade samt följas upp regelbundet 64. Rökstopp bör rekommenderas. Övriga levnadsvanor såsom hälsosamma matvanor och regelbunden fysisk aktivitet är också viktigt för att minska risken för hjärt-kärlsjukdom, förbättra livskvaliteten och lindra symtom.
Prefibrotisk myelofibros (pre-MF)
Rekommendationer
- Vid asymtomatisk sjukdom: observation. (+)
- Vid sjukdomssymtom eller kraftig myeloproliferativ sjukdom: IFN-alfa eller HU. (+)
- Vid högrisk IPSET score och/eller hjärt-kärlsjukdom: ASA. (+)
Prefibrotisk myelofibros (pre-MF) är sedan 2016 en egen sjukdomsentitet i WHO-klassifikationen. Jämfört med ET har pre-MF-patienter mer uttalade besvär med blödning, vilket åtminstone till en del är kopplat till trombocytos och CALR-mutation 84. Vidare ses en ökad risk att transformera till MF och akut myelotisk leukemi (AML) jämfört med ET 98. Långtidsprognosen vid pre-MF är trots detta i de allra flesta fall god.
Eftersom prospektiva behandlingsstudier saknas, baseras rekommendationerna på retrospektiva fallserier 99. För asymtomatiska patienter rekommenderas observation. För symtomatiska patienter samt för patienter med kraftig myeloproliferativ sjukdomsbild rekommenderas cytoreduktiv behandling med pegylerat interferon eller hydroxyurea. IPSET score 69 kan användas för att prediktera trombotiska händelser vid pre-PMF 100 där ingående parametrar är ålder >60 år, kardiovaskulära riskfaktorer, tidigare trombos och JAK2 mutation. Frånsett att patienter med pre-PMF tidigare inkluderats i behandlingsstudier vid ET saknas prospektiva studier avseende behandling med trombocythämmare. För patienter med hög risk IPSET score och/eller etablerad hjärt-kärlsjukdom bör ASA ges iakttagande den ökade blödningsrisken vid pre-MF.
Vid MF-liknande klinik med splenomegali och/eller anemi kan riskvärdering enligt IPSET ge vägledning. Vid symtomgivande splenomegali eller konstitutionella symtom kan ruxolitinib provas så som vid MF. I den ovanliga situationen med pre-PMF med högriskkriterier enligt IPSET är medianöverlevnaden under 5 år jämförbar med överlevnaden vid högrisk MF och i denna grupp bör allogen stamcellstranplantion övervägas som behandlingsval 101 men i övriga fall är långtidsprognosen god och generellt bör allo-SCT enbart övervägas vid tecken till MF eller vid leukemisk transformation.
Myelofibros (MF)
Rekommendationer för prognosticering
- Riskstratifiering bör utföras på samtliga patienter med myelofibros.
- Validerade riskstratifieringsmodeller för prognostisering:
- DIPSS: Vid diagnos och uppföljning (PMF)
- MIPSS-70, MIPSS-70+ v2.0: För patienter £70 år (PMF)
- MYSEC-PM: För post-PV och post-ET MF
- Dynamisk riskstratifiering rekommenderas vid förändring av sjukdomsbild eller behandlingssvar.
Rekommendationer för behandling
- Primär och sekundär myelofibros behandlas enligt samma principer.
Lågrisk myelofibros:
- Asymtomatisk patient: Observation utan aktiv behandling.
- Symtomatisk patient
- Ruxolitinib: Vid symtomgivande splenomegali och/eller konstitutionella symtom (++).
- Pegylerat IFN-alfa: Vid proliferativ sjukdom och låg fibrosgrad (+).
- Hydroxyurea: Vid proliferativ sjukdom för cytoreduktion.
- Allogen SCT: Bör övervägas för intermediärrisk-1 med högriskkriterier (++)
Högrisk myelofibros:
- Allogen SCT: Rekommenderas om ålder och samsjuklighet tillåter. (+++)
- JAK-hämmare: Vid symtomgivande splenomegali och / eller konstitutionella symtom (++++).
Mål med behandling
Behandling av myelofibros (MF) syftar till att bota sjukdomen i utvalda patientgrupper genom allogen stamcellstransplantation (allo-SCT), minska MF-relaterade symtom (anemi, splenomegali, konstitutionella symtom) samt minska risken för komplikationer (trombos, blödning, transformation). Behandlingen styrs i hög grad av riskstratifiering, MF-relaterade symtom och samsjuklighet.
Patienter med diagnosticerad myelofibros bör i ett tidigt skede diskuteras med regional eller nationell expertis, och frågan om allogen stamcellstransplantation bör då lyftas.
Primär och sekundär myelofibros behandlas enligt samma principer baserade på riskstratifiering och symtom.
Prognostiska modeller för PMF och post-PV/post-ET MF
Sjukdomsförloppet vid PMF och post-PV/post-ET myelofibros varierar betydligt mellan patienter. Därför rekommenderas att samtliga patienter med MF genomgår en riskstratifiering. Risknivån, i kombination med symtombild (eg. splenomegali och konstitutionella symtom) samt samsjuklighet, ligger till grund för prognos och stöd för behandlingsbeslut, inklusive ställningstagande till allo-SCT. Inga modeller kan dock för närvarande förutsäga vilka patienter som gynnas av specifika terapier. Regelbunden omvärdering av prognostiska faktorer rekommenderas, framför allt vid kliniska förändringar eller misstanke om sjukdomsprogression.
DIPSS
Dynamic International Prognostic Scoring System (DIPSS) är en central modell för riskstratifiering vid myelofibros och möjliggör en dynamisk bedömning av risknivå under hela sjukdomsförloppet [107]. DIPSS baseras på fem kliniska faktorer: ålder >65 år, hemoglobinnivå <100 g/L, leukocytvärde ≥25 x 10⁹/L, cirkulerande blaster ≥1 % samt förekomst av konstitutionella symtom (feber, viktnedgång >10 % senaste 6 månaderna, nattliga svettningar, feber >37,5°C utan klar orsak). Hemoglobinnivå <100 g/L ges en högre viktning (2 poäng) jämfört med övriga faktorer (1 poäng vardera). Tabell 3.
En ytterligare vidareutveckling av DIPSS är DIPSS-plus, där tre tilläggsfaktorer inkluderas: ogynnsam karyotyp, trombocytopeni (trombocytvärde <100 x 10⁹/L) och behov av erytrocyttransfusioner 102.
DIPSS delar in patienter i fyra riskkategorier: lågrisk, intermediärrisk-1, intermediärrisk-2 och högrisk. Medianöverlevnaden är betydligt kortare för patienter i högriskgruppen (18 månader för DIPSS) jämfört med lågriskgruppen (ej uppnådd medianöverlevnad för DIPSS). Tabell 4.
Tabell 3. Prognostiska scoringsystemet DIPSS.
|
Riskfaktor |
DIPSS |
|
Ålder > 65 år |
1 |
|
Konstitutionella symtom (feber, uttalade svettningar, viktminskning) |
1 |
|
Hemoglobin <100 g/L |
2 |
|
Leukocyter ≥ 25 × 109/L |
1 |
|
Cirkulerande blaster ≥ 1 % |
1 |
Tabell 4. Riskscore och medianöverlevnad i månader.
|
Riskgrupp |
DIPSS |
Medianöverlevnad (år från diagnos |
|
Lågrisk |
0 |
Ej uppnådd |
|
Intermediärrisk-1 |
1–2 |
14.2 |
|
Intermediärrisk-2 |
3–4 |
4 |
|
Högrisk |
≥ 4 |
1.5 |
Länk till DIPSS: https://qxmd.com/calculate/calculator_187/dipss-prognosis-in-myelofibrosis. Riskscore och medelöverlevnad i månader.
MIPPS-70 och MIPSS-70+ Version 2.0
För patienter ≤70 år har mer avancerade riskmodeller utvecklats, MIPSS-70 (Mutation-Enhanced IPSS) och MIPSS-70+ v2.0. Dessa modeller inkluderar molekylära och cytogenetiska faktorer och är särskilt användbara för att identifiera patienter som kan vara aktuella för allogen stamcellstransplantation. MIPSS-70 kombinerar kliniska faktorer, molekylära faktorer inklusive inklusive mutationer i ”high molcular risk” (HMR) gener. MIPSS-70+ v2.0 är en vidareutvecklad version där cytogenetiska faktorer och nya viktningsjusteringar för vissa kliniska variabler har lagts till. MIPSS-70 delar in patienter i tre riskkategorier (låg, intermediär och hög), medan MIPSS-70+ v2.0 har fem riskgrupper (very low, low, intermediate, high och very high). Även här är skillnaderna i överlevnad mellan grupperna stora.
High Molecular Risk (HMR) mutationer definieras som mutationer i gener som är förknippade med sämre prognos och ökad risk för leukemisk transformation och omfattar ASXL1, SRSF2, EZH2, IDH1 och IDH2. Förekomsten av fler än en HMR-mutation är kopplad till en särskilt dålig prognos. Utöver dessa har mutationer i TP53, U2AF1, RUNX1, CBL, NRAS och KRAS visat sig påverka prognosen negativt. Cytogenetiska förändringar förekommer hos ungefär 40 % av MF patienterna. Förekomst av inv(3), -5/5q-, -7/7q- +8, 11q23 och 12p−, i(17q) samt komplex karyotyp (definierat som > 2 avvikelser i karyotypen) är kopplade till sämre överlevnad och högre risk för leukemisk transformation. CALR-mutationer är förknippade med en bättre prognos jämfört med JAK2- och MPL-mutationer, men detta fördelaktiga utfall är framför allt begränsat till typ 1 och typ 1-liknande mutationer.
|
Mutation-enhanced IPSS (MIPSS) |
|
|
Prognostisk variabel |
Poäng |
|
Hemoglobin <100 g/L |
1 |
|
Leukocyter >25 x 109/L |
2 |
|
Trombocyter <100 x 109/L |
2 |
|
Cirkulerande blaster ≥2 % |
1 |
|
Fibrosgrad ≥2 I benmärg |
1 |
|
Konstitutionella symtom |
1 |
|
CALR typ 1-omuterad genotyp |
1 |
|
HMR mutationer: ASXL1, EZH2, SRSF2, IDH1/2 |
1 |
|
≥2 HMR mutationer |
2 |
|
Riskgrupp |
Poäng |
Medianöverlevnad |
|
Low |
0–1 |
Ej uppnådd |
|
Intermediate |
2–4 |
6.3 |
|
High |
≥5 |
3.1 |
|
Mutation and karyotype enhanced IPSS (MIPSS-70+ v2.0) |
|
|
Prognostisk variabel |
Poäng |
|
Svår anemi (Hemoglobin <80 g/L för kvinnor och <90 g/L för män) |
2 |
|
Medelsvår anemi (Hemoglobin 80 - 99 g/L för kvinnor och 90-109 g/L för män) |
1 |
|
Cirkulerande blaster ≥2 % |
1 |
|
Konstitutionella symtom |
2 |
|
Frånvaro av CALR typ 1 mutation |
2 |
|
HMR mutationer: ASXL1, EZH2, SRSF2, U2AF1 Q157 or IDH1/2 |
2 |
|
≥2 HMR mutationer |
3 |
|
Ofördelaktig karyotyp |
3 |
|
Very-high-risk (VHR) karyotyp |
4 |
|
Riskgrupp |
Poäng |
Medianöverlevnad (år) |
|
Very low |
0 |
Ej uppnådd |
|
Low |
1–2 |
16.4 |
|
Intermediate |
3–4 |
7.7 |
|
High |
5–8 |
4.1 |
|
Very high |
≥9 |
1.8 |
Ett onlineverktyg för MIPSS-70 och MIPSS-70+ Version 2.0 finns på http://www.mipss70score.it/.
MYSEC-PM: Prognostisk modell för sekundär myelofibros.
För patienter med post-PV och post-ET myelofibros används MYSEC-PM (Myelofibrosis Secondary to PV/ET Prognostic Model) för riskstratifiering. Modellen väger in kliniska och molekylära faktorer och identifierar fyra riskgrupper med signifikanta skillnader i överlevnad 103.
|
Myelofibrosis secondary to PV and ET-prognostic model (MYSEC-PM) |
|
|
Prognostisk variabel |
Poäng |
|
Ålder vid diagnos |
0.15 x ålder |
|
Hemoglobin <110 g/L |
2 |
|
Cirkulerande blaster |
2 |
|
Frånvaro av CALR typ 1 mutation |
2 |
|
Trombocyter <150x109/L |
1 |
|
Konstitutionella symtom |
1 |
|
Riskgrupp |
Poäng |
Medianöverlevnad (år) |
|
Low |
<11 |
Ej uppnådd |
|
Intermediate-1 |
11-13 |
9.3 |
|
Intermediate -2 |
14-16 |
4.4 |
|
High |
>16 |
2.0 |
Länk till MYSEC: http://www.mysec-pm.eu
Länk till samtliga ovanstående riskmodeller finns att tillgå: https://pmfscorescalculator.com/
Övriga riskstratiferingssystem
Andra prognostiska verktyg för MF inkluderar RR6-modellen och Myelofibrosis Transplant Scoring System (MTSS). RR6-modellen förutsäger överlevnad vid behandling med ruxolitinib efter sex månader, baserat på förändringar i mjältstorlek, dosintensitet och transfusionsbehov 104. MTSS är ett verktyg för att förutsäga överlevnad efter allo-HSCT och kombinerar faktorer såsom patientens ålder, hematologiska parametrar, molekylära faktorer, fysisk status och grad av HLA-matchning 105.
Skattning av symtombörda
Ett flertal validerade verktyg har utvecklats för att objektivt mäta symtombördan vid MPN. De mest etablerade verktygen inkluderar MF Symptom Assessment Form (MF-SAF), MPN Symptom Assessment Form (MPN-SAF) och MPN-SAF Total Symptom Score (MPN-SAF TSS, även kallad MPN-10) 106107.
Behandling med JAK hämmare
Ruxolitinib
Ruxolitinib är en JAK1/JAK2-hämmare som verkar genom att hämma JAK/STAT-signaleringen. Den är godkänd för behandling av primär eller sekundär myelofibros (post-PV MF och post-ET MF) och rekommenderas för patienter med intermediär-2 och högrisk MF. Ruxolitinib kan också övervägas vid symtomatisk splenomegali och/eller konstitutionella symtom hos patienter med intermediär-1 MF men saknar läkemedelssubvention för denna indikation.
Kliniska studier (COMFORT-1 och COMFORT-II) visar en signifikant minskning av mjältvolym och förbättring av symtom 108109110. Långtidsdata indikerar förlängd överlevnad för patienter som behandlas med ruxolitinib 111, dock har evidensstyrkan ifrågasatts 2.
JUMP-studien som även inkluderat patienter med intermediärrisk-1 visar att denna riskgrupp har en jämförbar säkerhetsprofil och likvärdig effekt av ruxolitinib jämfört med hela studiepopulationen 110.
Startdosen av Ruxolitinib baseras på trombocyttal enligt FASS. Vid uttalad anemi bör en lägre initialdos övervägas. Dosen titreras sedan utifrån behandlingens effekt och tolerans. Behandlingseffekten på konstitutionella symtom kan ofta noteras inom dagar medan effekten på mjältstorleken kan dröja varför definitiv utvärdering av behandlingseffekten rekommenderas först efter 6 månader. Snabb utsättning av ruxolitinib bör undvikas då det kan leda till utsättningssymtom.
Hematologisk toxicitet i form av anemi och trombocytopeni är vanligt förkommande i tidigt skede av behandlingen, men kan i vissa fall förbättras över tid. Dosökning bör därför genomföras långsamt, med intervall om (2–)4 veckors. Det bör dock understrykas att för flertalet patienter kvarstår de initiala cytopenierna utan nämnvärd förbättring.
Ruxolitinb är associerad med en ökad risk för opportunistiska infektioner och virusreaktiveringar. Screening för hepatit B och C, samt HIV och i utvalda fall tuberkulos rekommenderas före behandlingsstart. Hos patienter med hög risk för herpes zoster-reaktivering kan antiviral profylax övervägas.
En ökad risk att utveckla hudcancer under behandling med ruxolitinib har rapporterats och då särskilt hudcancer av icke-melanomtyp. Den exakta mekanismen bakom denna risk är inte fullt klarlagd, men immunmodulerande effekter av JAK-hämning tros spela en roll 112113114. Regelbunden bedömning av huden bör övervägas hos utvalda högriskpatienter, såsom de med en tidigare historia av hudcancer eller aktinisk keratos.
Fedratinib
Fedratinib är en selektiv JAK2 hämmare godkänd för behandling av primär eller sekundär myelofibros vid intermediär-2 och hög risk.
I JAKARTA-studien visade fedratinib signifikant reduktion av mjältvolymen och förbättring av symtom hos patienter med MF 115116. JAKARTA-2 visade även effektivitet hos patienter som tidigare behandlats med ruxolitinib 117. Läkemedelssubvention för denna indikation saknas dock.
Profylaktisk behandling med antiemetika rekommenderas de första behandlingsveckorna att hantera vanligt förekommande gastrointestinala biverkningar som illamående och diarré. Tiaminnivån bör kontrolleras före behandlingsstart och regelbundet övervakas för ställningstagande till tiamintillskott för att förebygga Wernickes encefalopati. Vid misstanke om brist, eller när regelbunden övervakning av nivåerna inte är möjlig, rekommenderas rutinmässig substitution med tiamin.
Försiktighet bör iakttas vid övergång från ruxolitinib till fedratinib för att undvika en utsättningsreaktion. Övergång mellan dessa läkemedel bör diskuteras med regionalt MPN-ansvarig hematolog.
Momelotinib
Momelotinib är en selektiv hämmare av JAK1/2 och AVCR1, godkänd för behandling av primär eller sekundär myelofibros hos patienter som har måttlig till svår anemi. Läkemedlet ingår i läkemedelsförmånen för patienter med intermediär-2 eller högrisk myelofibros. Godkännandet baserades på resultat från fas III-studierna MOMENTUM 118, SIMPLIFY-1 119 och SIMPLIFY-2 120. Genom att hämma ACVR1 minskar momelotinib uttrycket av hepcidin, vilket underlättar för erytropoesen och ger fördelar vid anemi.
Den rekommenderade startdosen är 200 mg dagligen, oberoende av trombocytvärdet. Momelotinib har i kliniska studier visats vara effektivt för att minska splenomegali och symtom relaterade till myelofibros, och kan övervägas för behandling av patienter med intermediär-II och högrisk myelofibros vid anemi.
Andra läkemedel vid myelofibros
Hydroxyurea
Hydroxyurea är en antimetabolit som verkar genom att hämma DNA-syntes. Läkemedlet är användbart i situationer där det primära målet med behandlingen är att kontrollera proliferativa blodvärden, ofta i de tidigare stadierna av sjukdomen. Vissa patienter med splenomegali och symtomgivans sjukdom kan dra nytta av behandlingen, men ihållande behandlingssvar är sällsynt. Behandlingen anpassas individuellt (0,5g/2d – 2g/d) baserat på patients blodvärden och tolerans. Läkemedelsincuderad anemi kan begränsa cytoreduktivt aktiv dos och regelbunden monitorering av blodvärden är nödvändig. HU kan kombineras med interferon-alfa och även ruxolitinib 121.
Interferon-alfa
Interferon-alfa (IFN-alfa) är en immunmodulerande behandling med dokumenterad effekt vid tidig myelofibros, särskilt hos patienter proliferativ blodbild och låg fibrosgrad 54122123124125126127. Kliniska studier visar att IFN-alfa utöver att uppnå hematologisk kontroll även kan minska graden av benmärgsfibros, särskilt hos patienter med låg initial fibrosgrad 126127. Behandlingsnyttan är emellertid begränsad hos patienter med mer avancerad fibros, där biverkningar ofta blir betydande.
I Sverige används primärt pegylerat interferon-alfa (peginterferon alfa-2a), som har en lång tradition som cytoreduktiv behandling vid myeloproliferativa sjukdomar, inklusive tidig myelofibros. Det är också det enda rekommenderade alternativet för gravida patienter, även om läkemedlet saknar en specifik indikation i FASS.
Peginterferon alfa-2a administreras subkutant, vanligen som en veckodos om 45 - 90 mg eller varannan vecka med en dos om 135 mg. Behandlingen bör påbörjas med låg dos och titreras baserat på klinisk respons och tolerans. Regelbunden monitorering av lever- och tyroideafunktionen är nödvändig. Efter att ett stabilt behandlingssvar har uppnåtts kan dosen ofta reduceras över tid för att minimera biverkningar.
Vanliga biverkningarna inkluderar influensaliknande symtom. Psykiatriska biverkningar, såsom depression, samt autoimmuna tillstånd kan uppstå och bör föranleda en omvärdering av fortsatt behandling. Hos patienter med anamnes på autoimmun sjukdom eller psykiatriska tillstånd bör särskild försiktighet iakttas vid insättning av behandlingen.
Trombocytaggregationhämmare
Vid bakomliggande riskfaktorer för hjärt-kärlsjukdom eller etablerad hjärt-kärlsjukdom bör ASA ges iakttagande den ökade blödningsrisken som kan råda vid myelofibros. Dubbelterapi med ASA och antikoagulantia bör endast ges vid specifika behandlingsskäl för annan diagnos.
Behandling av MF-associerad anemi
Behandling av anemi vid MF rekommenderas vid hemoglobinnivåer <100 g/l, eller <110 g/l vid symtom 2. Innan behandling påbörjas bör bakomliggande orsaker till anemi uteslutas, inklusive blödning, järn-, vitamin B12- och folatbrist samt hemolys.
Erytropeosstimulerande läkemedel (ESA), såsom rekombinant humant EPO och darbepoietin alfa, kan förbättra anemi hos patienter med MF. Faktorer som har visat sig vara associerade med en bättre behandlingsrespons är låga serum-EPO nivåer (<125 mU/L), mindre mjältstorlek och lågt behov av erytrocyttransfusioner 128. Rekommenderad startdosering för rekombinant humant EPO är 30 000 I.E. per vecka och för darbepoietin alfa 150–300 µg/vecka. ESA kan även kombineras med ruxolitinib 129.
Vid myelofibrosorsakad anemi kan kortikosteroider vara ett behandlingsalternativ men svaret är ofta kort 130.
Erytrocyttransfusioner är indicerade för att snabbt häva symtomgivande anemi eller vid terapisvikt på annan anemibehandling. Ett kroniskt transfusionsbehov är förknippat med dålig prognos och lägre livskvalitet 131.
Det finns för närvarande inga övertygande data som stödjer användning av keleringsbehandling vid MF 132. Därför rekommenderas kelering endast för patienter som är kandidater för allo-SCT 133134.
Splenektomi och strålning mot mjälten
Splenektomi och strålning mot mjälten bör bara användas i de få undantagsfall där andra behandlingsval eller deltagande i klinisk studie inte är aktuellt 2135136137138. Strålning mot mjälten kan på grund av hämmad hematopoes orsaka en uttalad och långdragen cytopen situation, och splenektomi innefattar i denna patientgrupp en hög risk för perioperativ dödlighet och sjuklighet 139.
Allogen stamcellstransplantation (allo-SCT)
Allogen stamcellstransplantation (allo-SCT) är den enda potentiellt botande behandlingen för myelofibros. Risken för transplantationsrelaterad mortalitet och morbiditet är dock betydlig med cirka 20–35 % transplantationsrelaterad mortalitet och en 5-årsrisk för återfall på cirka 20–30 % 140141. Från en svensk-norsk sammanställning av 139 patienter transplanterade 2009 - 2019 fann man en förbättrad överlevnad hos gruppen som blev transplanterade 2015-2019 (n=84) jämfört med 2009-2014 (n=55). Man fann en 3‑årsöverlevnad på 81 % jämfört med 66 %.
Femårsöverlevnaden efter allo-SCT ligger på cirka 50–67 % enligt EBMT. Överlevnaden hos äldre (> 55 år) är dock betydligt sämre än hos yngre (48 % versus 82 %). Patienter som svarar på behandling med JAK1/2-hämmare har en bättre överlevnad än patienter som inte svarar på ruxolitinib 142.
Patienter med primär myelofibros och intermediär-2 eller högrisk enligt DIPSS eller högrisk enligt MIPSS70 eller MIPSS70 plus bör transplanteras förutsatt att ålder och samsjuklighet tillåter. På samma sätt bör även patienter med sekundär myelofibros med intermediär2 eller högrisk enligt MYSEC-PM transplanteras 140141.
Man kan även använda DIPSS som en del av ett beslutsunderlag. Vid lägre riskscore får ett än mer individuellt ställningstagande göras där man kan väga in andra faktorer (10.4.10).
För att prediktera utfall av allo-SCT vid myelofibros kan Myelofibrosis Transplant Scoring System (MTSS) användas. Emellertid bygger data från retrospektiva jämförande studier som inte är validerade prospektivt 105143. Patienter med intermediärrisk-1 PMF (DIPSS) eller intermediärrisk MIPPS 70 eller 70plus är potentiella kandidater för allogen stamcellstransplantation, förutsatt att ålder och samsjuklighet tillåter (MTSS låg risk), om följande föreligger:
- Refraktär, transfusionsberoende anemi
- > 2 % blaster i perifert blod
- Högriskcytogenetik enligt DIPSS-plus
- Trippelnegativ MF, det vill säga frånvaro av mutation i JAK2V617F, CALR och MPL
- ASXL-1-mutation
- TP53
Beslutsprocessen bör diskuteras med den MPN-transplantationsansvariga i regionen.
Behandling med JAK-inhibitor rekommenderas inför transplantation hos patienter med konstitutionella symtom och/eller symtomatisk splenomegali 144. Hos patienter med minskad mjältstorlekt vid behandling ses en minskad risk för återfall samt förbättrad överlevnad.
Oklassificerbar MPN (MPN-U)
Oklassificerbar myeloproliferativ neoplasi (MPN-U) är en ovanlig subtyp av MPN som omfattar fall där de diagnostiska kriterierna för en specifik MPN-subtyp inte uppfylls eller som uppvisar egenskaper som överlappar med två eller flera subtyper 145146. De flesta fall är positiva för JAK2 V617F, CALR eller andar myeloida drivande mutationer.
Trombos, inklusive tromboser på atypiska lokaler såsom levernära tromboser, är en vanlig manifestation vid MPN-U och utgör inte sällan det första tecknet på sjukdomen före diagnos.
Historiskt har MPN-U utgjort cirka 10 % av de patienter som rapporterats i det nationella MPN-registret. Vid treårsuppföljning har cirka 45 % av dessa patienter omklassificerats till en specifik MPN-subtyp (interaktiv rapport MPN). För ett flertal av de inrapporterade patienterna saknas också tillräckligt diagnostiskt underlag, särskilt i form av benmärgsbiospi vilket är en nyckelkomponent i WHO:s kriterier för MPN-diagnostik. Avsaknaden av benmärgsbiopsi gör att dessa fall inte uppfyller WHO-kriterierna för MPN-U.. Registervalidering har också visat att flera fall, som initialt klassificerats som MPN-U, uppfyller kriterier för myelodysplastisk/myeloproliferativ neoplasi (MDS/MPN).
Eftersom MPN-U är en ovanlig och heterogen subtyp föreslås följande diagnostiska förhållningssätt i syfte att förbättra klassificeringen:
- Överväg MDK alternativt kontakt med regional MPN-representant.
- NGS (myeloid panel rekommenderas vid negativ MPN-genetik (JAK2, CALR och MPL) eller vid förekomst av i benmärg dysplastiska drag i erytropoes och granulopoes, eller vid monocytos.
- Ett dynamiskt förhållningssätt vid uppföljning där diagnosen bör omvärderas vid förändringar i den kliniska bilden.
Det finns idag inga vedertagna riktlinjer för behandling av MPN-U och behandlingen bör därför individualiseras och särskilt beakta trombosrisk, proliferativ blodbild och konstitutionella symtom 147. Vid misstanke om sjukdomsprogression bör ställning tas till förnyad diagnostisk utvärdering innefattande benmärgsbiopsi, molekylärgenetik och bedömning av mjältstorlek.