
Bilaga 1 Kvalitetsdokument för patologi
Patologins roll i den diagnosiska processen
Patologens bedömning av lesioner i binjuren är en sammansatt process som innefattar makroskopiska, mikroskopiska och molekylära analyser. Det finns en kliniskt accepterad gråzon när det gäller tumörernas malignitetspotential. Om det inte finns definitiva bevis på en malign tumör (såsom fjärrmetastaser) bygger patologens bedömning i stället på en sammanvägning av en rad ”riskkriterier”. Detta, i kombination med låga patientvolymer, gör att binjuretumörer bör diagnostiseras av ett mindre antal subspecialiserade patologer.
Klassificering av tumören
Binjuretumörer klassificeras enligt WHO 2022 [1, 2] Binjurebarkscancer, feokromocytom och paragangliom ska även stadieindelas enligt TNM (AJCC’s 8:e upplaga 2017) [3].
Utskärningsanvisningar
Biopsier mäts och bäddas i sin helhet. Operationspreparat (resektat) skärs ut enligt följande:
- För operationspreparat från binjure och paragangliom anges preparatvikt (gram), primärtumörens eller primärtumörernas lokalisation, storlek i tre dimensioner (millimeter) och avstånd till närmsta resektionsrand.
- Makroskopiskt anges eventuell multifokalitet, tumörens snittyta, färgton, eventuell nekros, synbara tecken på kapselbrott eller extraadrenal extension i angränsande fettvävnad, liksom direkt inväxt i vena cava inferior. För patienter med kända ärftliga syndrom (med konstitutionella mutationer i framför allt NF1, MEN2, VHL och/eller SDHx) bör stor noggrannhet fästas vid eventuell multifokalitet samt breddökning av märgskiktet (adrenomedullär hyperplasi, AMH), där binjuremärgen upptar mer än en tredjedel av binjurens totala tjocklek i tvärsnitt. Därtill bör förekomst av makroskopiskt synlig märgvävnad i binjurevingarna noteras, vilket också indikerar AMH.
- För binjurebarkslesioner är bedömning av antalet patologiskt synbara barknoduli viktig för att skilja mellan adenom och hyperplasi. Alla uppdrivningar i binjurebarken utöver huvudlesionen bör beskrivas och fraktioneras till mikroskopi. Kortisolproducerande lesioner tenderar att vara brandgula i färgtonen medan aldosteronproducerande noduli ofta uppvisar en klassisk kanariegul snittyta.
- Skivor tas från tumören eller tumörerna, närmaste resektionsrand och eventuella regionala lymfkörtlar. Storsnitt kan vara av värde vid större lesioner med multifokala expansioner mot kapseln, men delar av tumören bör finnas tillgänglig i konventionella kassetter för immunhistokemisk utredning.
Alla lymfkörtlar i vidhängande vävnad bäddas.
Analyser
Biopsier och utskurna vävnadsblock paraffinbäddas, snittas och färgas med hematoxylin-eosin. Malignitetsmisstänkta tumörer ska färgas immunhistokemiskt för att säkerställa diagnos och ursprung.
Malignitetsmisstänkta lesioner i binjurebark bör verifieras med antikroppar mot barkmarkörer, t.ex. melan A, inhibin-alfa, SF1 och kalretinin [4, 5]. Därtill bör åtminstone kromogranin-A undersökas för att utesluta feokromocytom. Synaptofysin är i detta avseende en olämplig färgning eftersom denna markör ofta är positiv även i barklesioner. Keratinmarkörer är generellt negativa i barktumörer. Ki-67 (MIB1) bör undersökas för att ange proliferationsindex. Endotelcellsmarkörer (CD31, CD34 och/eller ERG) är värdefulla vid utredning av misstänkta foci med kärlinvasion, och Van Gieson-histokemi kan vara till hjälp vid bedömning av kapselbrott.
För feokromocytom och parangangliom bör immunhistokemi inkludera chromogranin A, synaptofysin, CD56, S100 och Ki-67 (MIB1)[1, 6]. S100 används för att identifiera s.k. satellitceller/sustentacular cells, stödjeceller i utkanten av de klassiska Zellballen-strukturerna (tumörnästen). Observera att en undergrupp av pheochromocytom och paragangliom även kan färga in nukleärt för S100. Färgning mot tyrosin-hydroxylas (TH) är oftast positiv i funktionella pheochromocytom och sympatiska paragangliom. Även dopamin-β-hydroxylas är en användbar markör i sammanhanget, eftersom vissa icke-funktionella feokromocytom och sympatiska paragangliom kan sakna TH-immunoreaktivitet. Cytokeratin-färgningar är negativa i pheochromocytom och paragangliom. De keratin-positiva lesioner med neuroendokrin profil som ses i cauda equina benämns numera som ”cauda equina neuroendocrine tumor (cauda equina-NET) snarare än ”cauda equina-paragangliom” [1]. Dessa tumörer uppvisar ej GATA3-positivitet och är ej associerade med genetiska syndrom på samma sätt som konventionella paragangliom, vilket gör namnbytet logiskt. På samma sätt har entiteten ”gangliocytiskt paragangliom” som ofta uppstår i duodenum ersatts av termen ”composite gangliocytoma/neuroma and neuroendocrine tumor (CoGNET)”, då även denna tumör uppvisar keratinpositivitet inom den neuroendokrina komponenten, samt därtill färgar in negativt för GATA3 men ofta ses med inmärkning mot somatostatin, serotonin och/eller vasoaktiv intestinal polypeptid (VIP) [1].
Beroende på klinisk frågeställning, tumörlokalisation och mikroskopiska fynd, kan ytterligare immunhistokemiska analyser utföras. SDHB-immunohistokemi har visat sig korrelera till underliggande mutation i SHDx-isoformer, och kan därför vara värdefull vid utvärdering av framför allt paragangliom när man väljer fall för närmare genetisk utredning [7, 8]. Carbonic anhydrase IX (CAIX) kan även vara värdefull för att identifiera tumörer som uppvisar underliggande VHL-mutation, och Inhibin-alfa kan också övervägas för att detektera tumörer som har mutation i gener som kopplas till pseudohypoxi-klustret (med en ökad risk för framtida metastasering jämfört med tumörer som uppvisar mutationer i kinas-relaterade gener) [9, 10].
För ovanliga diagnoser (ganglieneurinom, myelolipom och schwannom) kan tilläggsanalyser med diagnos-specifika antikroppar nyttjas.
Vid metastatisk tumörsjukdom (förekomst av kromaffina celler utanför märg eller paraanglier) kan färgning för chromogranin A, synaptofysin, CD56 och GATA3 ge viss vägledning om metastaserande pheochromocytom. Observera att här finns en stor likhet i den aktuella profilen jämfört med andra neuroendokrina tumörer (NET), och så en biopsiverifierad metastas av pheochromocytom och paragangliom bör om möjligt även inkludera information om epitelial markör (AE1/AE3), CDX2, serotonin (tunntarms-NET), PDX1 (NET från pankreas ellerövre GI) och TTF1 (lunga, och thyroidea) [11]. Då keratinmarkörer är negativa i pheochromocytom och paragangliom kan ett positivt utfall indikera neuroendokrin tumör med annan primaritet än binjuremärgellerparaganglier.
Vid misstanke om metastas till binjuren från primärtumör i annat organ bör immunhistokemi dels inkludera ett mindre antal bark- och märgmarkörer enligt ovan för att säkert utesluta ursprung på platsen, och dels inrikta sig på att verifiera primärtumörens ursprung. Vanligast är metastaserande njurcancer av klarcellig typ samt adenocarcinom från lunga så för att täcka in allt används en panel bestående en eller flera njur- (EMA, vimentin, CD10, PAX8, RCC) samt lung-markörer (TTF1, CK7 Napsin A) vara heltäckande. Övriga metastaser färgas med sedvanligt organspecifika markörer som är kända enligt gängse litteratur.
I de fall laboratoriet saknar rekommenderade antikroppar bör fallet skickas för konsultation. Endokrinpatologer med tillgång till dessa färgningar finns tillgängliga på Karolinska Universitetssjukhuset i Solna och Akademiska sjukhuset i Uppsala.
Information i remissens svarsdel
Makroskopisk beskrivning
Makrobeskrivningen följer utskärningsanvisningarna ovan.
Mikroskopiutlåtande
Lesioner i binjurebark
Binjurebarkslesioner utgörs oftast av binjurebarksadenom eller binjurebarkshyperplasi, medan binjurebarkscancer är ovanlig. Distinktionen mellan benigna och maligna barktumörer är i många fall svår att göra, och i vissa fall kan man få ledtrådar via tumörens storlek (binjurebarkscancer nästan alltid >4 cm) och/eller förekomst av preoperativt avgjord radiologisk misstanke om tumörspridning (inväxt i vena cava inferior och/eller lokal lymfadenopati).
Ett mikroskopiutlåtande för binjurebarkslesioner lesioner ska innefatta de parametrar som krävs för att korrekt uppskatta av lesionens maligna potential enligt specifika Weiss-, Lin-Weiss-Bisceglia- eller Wieneke-kriterier. Enligt WHO 2022 kan även de så kallade Helsinki- och retikulin-algoritmerna ingå som hjälpmedel i bedömningen av binjurebarkstumörer, även om de som prognostiska verktyg är yngre och har färre validerande studier bakom sig [12, 13]. Nedan följer parametrar som oavsett klassifikationssystem är av värde vid utvärderingen av binjurebarkstumör:
- Förekomst av multicentricitet.
- En generell histologisk beskrivning av lesionen eller lesionernas utseende såsom växtmönster och cytologiska attribut där fördelningen mellan a) fascikulata-liknande celler med låg kärna:cytoplasma-kvot och lipidrik cytoplasma och b) reticularis-liknande eosinofila celler kan ge utslag på den s.k. Weiss-skalan. Vidare kommenteras eventuell förekomst av oxyfila celler, pleomorfism, mitostaloch äkta tumörnekros.
- Förekomst av tumörkapsel.
- Förekomst av kapselinfiltration.
- Förekomst av blodkärlsinvasion inom kapselplanoch sinusoidalt.
- Förekomst av extraadrenal extension (oftast fettväv).
- Weiss-klassifikation, Helsinki-algoritmen, retikulin-algoritmen, Lin-Weiss-Bisceglia-klassifikation (oxyfila tumörer).
- Wieneke-klassifikation (pediatriska tumörer).
- Förekomst av tumörväxt i resektionsytor.
- Relevanta patologiska förändringar i icke-tumoröst parenkym (adrenomedullär hyperplasi, inflammation etc.)
- Vid medföljande lymfkörtlar antal och status.
- I fall med lymfkörtelmetastaser, den största metastasens storlek, samt förekomst av periglandulär växt.
Stratifiering av malign potential i binjurebarkstumörer
Den ursprungliga versionen av Weiss-klassifikationen är enligt WHO 2022 den äldsta histologiska algoritm som rekommenderas vid bedömning av binjurebarkstumörer och deras maligna potential [2, 14]. Det finns öven en omarbetad version (”modified Weiss”) som inte har fått lika stort genomslag.
Om lesionen är onkocytär (>90% av tumörcellsandelen) används istället den alternativa klassifikationsmodellen enligt Lin-Weiss-Bisceglia, eftersom dessa tumörer, benigna som maligna, genom sin oxyfila karaktär alltid får tre poäng i Weiss-klassifikationen (för nukleär atypi, <25% klara celler och diffust växtmönster) [2, 15] Helsingfors-modellen (som även inkluderar proliferations-index med Ki-67-antikropp) samt retikulin-algoritmen (som använder sig av retikelfärgning) går att applicera på konventionella såväl som oxyfila tumörer. Även ovanliga subtyper av binjurebarkstumörer existerar, t. ex. myxoida varianter, men det är inte känt om dessa tumörer kan klassificeras via en konventionell Weiss-gradering.
Weiss-klassifikationen utgår från nio histologiska parametrar, där förekomst av varje enskild parameter ger ett poäng på Weiss-skalan (Tabell 1). Tre eller fler poäng indikerar malign potential.
Tabell 18. Den ursprungliga Weiss-klassifikationen [14]
|
Histologisk parameter |
Poäng |
|
Nukleär atypi (Fuhrman-grad III eller IV) |
1 poäng |
|
> 5 mitoser/50 high power field |
1 poäng |
|
Förekomst av atypiska mitoser |
1 poäng |
|
Klara/vakuolförande celler utgör < 25 % av tumören |
1 poäng |
|
Kompakt växtsätt (diffus arkitektur) i > 30 % av tumören* |
1 poäng |
|
Förekomst av tumörnekros |
1 poäng |
|
Venös invasion (enbart i eller utanför kapselplan) |
1 poäng |
|
Sinusoidal invasion (inom tumören) |
1 poäng |
|
Kapselinvasion |
1 poäng |
Summa 3 poäng eller mer indikerar malign potential.
* Kan faciliteras via retikelfärgning (Gordon-Sweet).
Lin-Weiss-Bisceglia-klassifikationen ska enbart användas om lesionen är onkocytär (definieras som >90% onkocytära celler enligt WHO 2022). Hit räknas inte adrenokortikala lesioner med antytt eosinofil cytoplasma (retikularis-liknande celler), utan cellerna måste uppvisa stor, bulkig och i varierande grad granulerad cytoplasma. Klassifikationen är uppbyggd kring ”major criteria” (översättes till ”huvudsakliga kriterier”) och ”minor critera” (översättes till ”mindre kriterier”) enligt nedan:
Tabell 19. Lin-Weiss-Bisceglia-klassifikationen för oxyfila adrenokortikala tumörer [15]
|
Huvudsakliga kriterier |
|
> 5 mitoser/50 high power fields |
|
Förekomst av atypiska mitoser |
|
Venös invasion (enbart i eller utanför kapselplan) |
|
Mindre kriterier |
|
Tumör > 10 cm eller > 200 gram |
|
Förekomst av tumörnekros |
|
Sinusoidal invasion (inom tumören) |
|
Kapselinvasion |
Förekomst av minst ett av de huvudsakliga kriterierna är förenligt med onkocytär adrenokortikal cancer. Om enbart mindre kriterier (ett eller flera) uppfylls utgörs tumören av en onkocytär adrenokortikal tumör med oklar malign potential.
Wieneke-klassifikationen är framtagen för bedömning av malignitetspotential avseende binjurebarkstumörer hos pediatriska patienter, då den ursprungliga Weiss-algoritmen leder till en överdiagnostik av binjurebarkscancer i pediatriska populationer, visat i studier där tumörer med benigna förlopp och lång uppföljningstid erhållit falskt för höga Weiss-poäng.
Tabell 20. Wieneke-klassifikationen för pediatriska binjurebarkstumörer [16] -Weiss
|
Parameter |
Poäng |
|
Tumörvikt > 400 g |
1p |
|
Tumörstorlek > 10,5 cm |
1p |
|
Periadrenal extension eller inväxt i intilliggande organ |
1 p |
|
Invasion av v. cava |
1 p |
|
Kärlinvasion |
1 p |
|
Kapselinvasion |
1 p |
|
Tumörnekros |
1 p |
|
> 15 mitoser per 4 mm2 |
1 p |
|
Atypiska mitoser |
1 p |
|
Total poäng |
9 p |
Malignt beteende: ≥ 4 p
Okänd malignitetspotential: 3 p
Benignt beteende: ≤ 2 p
Helsinki-algoritmen är en viktad poängskala där poäng erhålls för mitoser, nekros och Ki-67-index enligt en specifik formel. Denna modell har reproducerats av oberoende grupper, och är enligt WHO 2022 en vedertagen metod för att identifiera binjurebarkscancer [12]. Retikulin-klassifikationen bygger på användandet av retikelfärgning via Gordon Sweet, där ett stört retikelnätverk i kombination med antingen förhöjt mitostal, nekros och/eller vaskulär invasion renderar en cancerdiagnos [13]. Det bör nämnas att medlemmar av endokrin-KVAST har begränsad erfarenhet av dessa två algoritmer, och dess betydelse för diagnostiken i Sverige är ej beprövad.
Fastställd binjurebarkscancer ska graderas som låg- eller höggradig enligt WHO 2022, och denna gradering bygger på mitosräkning. Låggradiga carcinom uppvisar ≤ 20 mitoser per 10 mm2, medan höggradiga carcinom ses med >20 mitoser per 10 mm2 [2].
Distinktion mellan binjurebarksadenom och hyperplasi
WHO 2022 har uppdaterat nomenklatur och kategorisering av adrenokortikala noduli, och schematiska översikter över dessa tillstånd finns återgivna som Figur 1 och Figur 2. I korthet kan nämnas att binjurelesioner med en preoperativt fastställd lateraliserad hormonproduktion (oftast aldosteron- eller kortisolproduktion) samt låg Weiss-poäng, kan om lateraliseringen är korrekt, i princip endast utgöras av ett hormonproducerande binjurebarksadenom eller multifokala adrenokortikala nodulära tillstånd (Figur 1-2). Att särskilja dessa tillstånd har en prognostisk betydelse, eftersom adenompatienter oftast botas med adrenalektomi och avskrivs från uppföljning, medan patienter med multifokala noduli kan uppvisa en risk för framtida återfall i den kontralaterala binjuren och därför ska följas [11]. Detta innebär att en felaktig diagnos kan skapa onödig uppföljning av botade patienter, alternativt prematur avskrivning av patienter som behöver vidare uppföljning.
Fynd av ett solitärt nodulus > cm indikerar vanligtvis adenom, medan den heterogena gruppen med multinodulär sjukdom kräver multifokala lesioner med likartad morfologi. Detta innebär att den makroskopiska bedömningen avgörr differentialdiagnostiken. Oavsett diagnos ser lesionerna ofta likartade ut rent histologiskt. De kan ses med eller utan omgivande kapsel, men är oftast avgränsade från övrig binjurebark av ett någorlunda nodulärt arrangemang av celler, inte sällan fascikulata-liknande celler med låg kärna:cytoplasma-kvot och lipidrik cytoplasma samt tillblandning av reticularis-liknande eosinofila celler.
Histologiskt kan det vara svårt att skilja mellan binjurebarksadenom och multifokal nodulär sjukdom. Kommersiellt tillgängliga antikroppar riktade mot CYP11B1 och CYP11B2 (enzymer som katalyserar bildandet av kortisol- respektive aldosteron) har tilläggsvärde för att skilja mellan dessa tillstånd när det preoperativt finns en lateraliserad hormonproduktion. I studier har förmodade adenom ibland visat sig vara icke-producerande eller kortisolproducerande. I stället har den sanna aldosteronproduktionen härstammat från flera minimala intilliggande noduli. Således rekommenderas denna funktionella immunhistokemi av WHO 2022 vid utvärderingen av adrenokortikala lesioner [2, 17].
För icke-producerande och kortisol-producerande lesioner i binjurebarken rekommenderas enligt WHO 2022 en indelning som återges i Figur 1. Solitära förändringar >1 cm benämns binjurebarksadenom eller -carcinom beroende på eventuella maligna attribut. Om storleken understiger 1 cm så benämns ett eller flera icke-producerande noduli som ”sporadisk nodulär adrenokortikal sjukdom” [2]. Denna entitet ses i alla åldrar, och kan vara unilateral eller bilateral. Om förändringarna är multipla och producerar kortisol så klassas dessa som antingen ”bilateral mikronodulär adrenokortikal sjukdom” (om noduli <1 cm) och ”bilateral makronodulär adrenokortikal sjukdom” (om noduli >1 cm) [2] . Dessa noduli ses då med positivitet för CYP11B1. Den bilaterala mikronodulära adrenokortikala entiteten delas in ytterligare i ”primär pigmenterad adrenokortikal sjukdom” och ”isolerad mikronodulär adrenokortikal sjukdom” enligt fastslagna kriterier i Figur 1. Kännetecknande för samtliga bilaterala tillstånd med kortisolproduktion är att de 1) uppvisar en reell recidivrisk på kontralateral binjure och således måste följas kliniskt samt 2) ofta uppvisar patogena varianter i olika gener som predisponerar för utvecklandet av ytterligare barknoduli i kvarvarande binjure [2, 5].
Figur 1. Översikt av adrenokortikal nodulär sjukdom (icke-producerande eller kortisol-producerande). Sjd; sjukdom. Skapad med BioRender.com. Modifierad från [5].
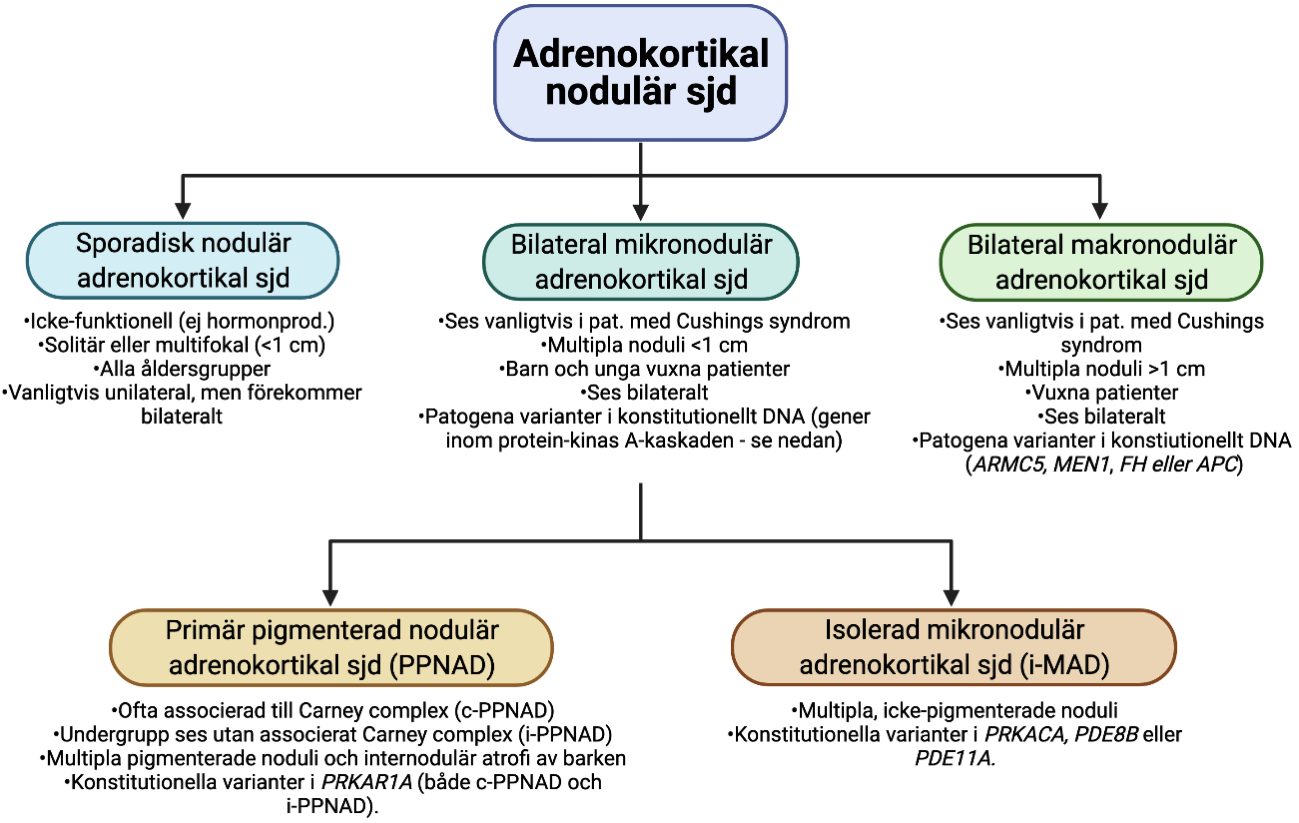
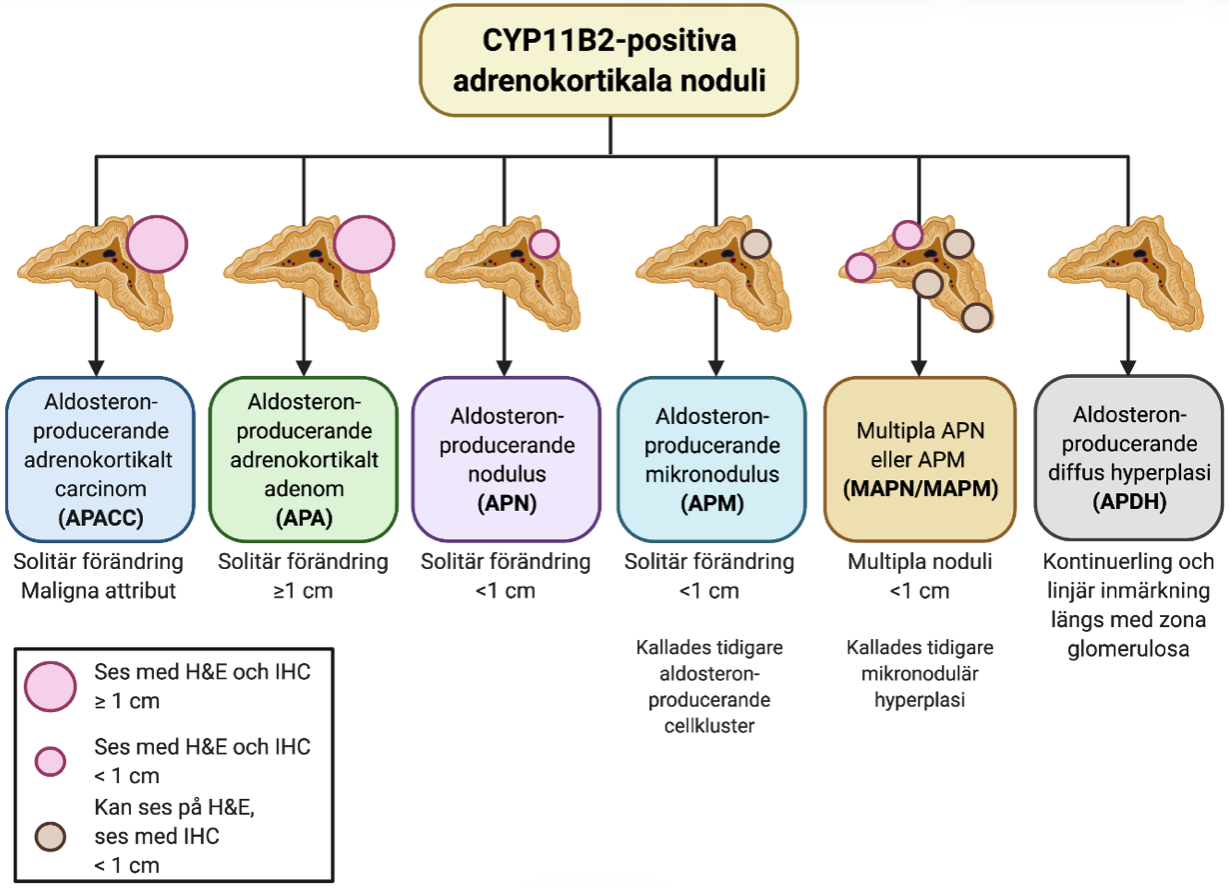
För patienter med primär hyperaldosteronism och ett eller flera noduli inom binjurebarken rekommenderas CYP11B2 som immunhistokemisk tilläggsmarkör vid utvärderingen av dessa förändringar (Figur 2) [2, 17]. För förändringar <1 cm skiljer man på solitära förändringar som detekteras i rutinfärgningar (hematoxylin-eosin) (”aldosteronproducerande nodulus; APN”) från förändringar som enbart noteras i immunhistokemisk färgning mot CYP11B2 (”aldosteronproducerande mikronodulus; APMN”) [5]. Om dessa noduli ses multifokala så används i stället benämningarna ”multipla APN; MAPN” och ”multipla APMN; MAPMN” [5]. Om en binjure uppvisar multifokala noduli så föreligger en betydande recidivrisk på kontralateral binjure. Aldosteronproducerande diffus hyperplasi kännetecknas av en kontinuerlig, band-formad CYP11B2-inmärkning inom zona glomerulosa.
Figur 2. Översikt av aldosteron-producerande adrenokortikala ooduli som färgar in för CYP11B2 (aldosteronsyntas). H&E; hematoxylin-eosin, IHC; immunhistokemi. Skapad med BioRender.com. Modifierad från [11].
Feokromocytom samt abdominala paragangliom
Alla feokromocytom och abdominella paragangliom är potentiellt maligna enligt WHO 2022 [1]. Det enda säkra kriteriet för malignitet är metastasering, och rekommendationen är att klassificera tumörer i kategorierna metastatiskt respektive icke-metastatiskt feochromocytom eller paragangliom. Återfall på samma plats efter tidigare kirurgi av ett abdominellt paragangliom kan dock rent tekniskt utgöra asynkron, multifokal tumörbörda till följd av nedärvd mutation i predisponerande gen, och det bör inte i sig användas som bevis för en malign sjukdom.
WHO 2022 skiljer mellan sympatiska (i huvudsak abdominella) och parasympatiska (i huvudsak huvud- och hals-) paragangliom. Det bör noteras att parasympatiska paragangliom eller huvud-halsparagangliom inte stadieindelas enligt TNM-systemet, och den histopatologiska riskbedömningen beskriven nedan ska inte tillämpas på dessa tumörer.
Två histologiska risk-system finns vilka enligt vissa studier kan predicera malignt beteende, men inte enligt andra [18-22]. WHO 2022 tar inte ställning för någon av dessa algoritmer, men vill inte heller avråda från implementering om ett center starkt önskar detta [1]. WHO listar därtill ett antal risk-kriterier som hämtas från respektive scoring-systems original-publikationer. Det är lämpligt att i PAD notera dessa histopatologiska riskkriterier inför värdering av tumörens riskprofil på MDK även om dessa faktorer var för sig har suboptimala prediktiva värden:
- Invasion (av kärl, kapsel eller periadrenal mjukvävnad)
- Växtmönster (oregelbundna, förstorade och konfluerande cellnästen)
- Cellbild (spolcellighet, småcellighet, hög cellulär densitet, monotoni, pleomorfism)
- Nekros (fokal eller konfluerande, komedonekros)
- Proliferativ aktivitet (förhöjt mitostal, atypiska mitoser, förhöjt Ki-67-index)
Det huvudsakliga klassifikationssystemet i WHO för feokromocytom, PASS (Pheokromocytoma of the Adrenal Gland Scaled Score), tar upp samtliga av dessa faktorer förutom småcelligheten, och har därtill en ytterligare faktor att beakta, nämligen nukleär hyperkromasi (se detaljerat schema nedan). PASS publicerades för första gången 2002 och har sedan dess reproducerats vetenskapligt i flera studier [18, 19, 22]. Denna algoritm bygger på identifikation av 12 histologiska parametrar, som var och en ger 1 eller 2 poäng. Summan 4 poäng eller mer indikerar potential för aggressivt kliniskt förlopp i form av framtida metastasering. PASS-modellen har bristande reproducerbarhet i vissa studier, främst på grund av dålig överensstämmelse mellan olika patologer. Detta tros bero på att originalpublikationen är något vag i definitionen av de olika innefattande parametrarna, och därmed lämnar utrymme för subjektiv bedömning. Värdet av en poängsättning är i första hand kopplat till PASS-algoritmens höga negativa prediktiva värde, där en låg PASS-poäng indikerar feokromocytom utan risk för framtida metastasering
År 2014 kom klassifikationssystemet; GAPP (The Grading system for Adrenal Pheochromocytoma and Paraganglioma) [21]. GAPP är ett derivat av PASS med tillägg i form av proliferationsindex samt biokemi, och de histologiska parametrar som ingår i GAPP finns även angivna i PASS.
Paragangliom som är associerade till SDHB-mutation har hög risk för framtida metastasering, och upp till en tredjedel av patienterna med denna mutation uppvisar spridd sjukdom. SDHB-immunhistokemi korrelerar till underliggande mutationsstatus och avsaknad av immunoreaktivitet kan indikera underliggande mutation i denna gen, alternativt en annan gen inom SDHx-familjen (SDHA, SDHC, SDHD) [8]. Carbonic anhydrase IX (CAIX) ses positiv i fall med underliggande VHL-mutation, och Inhibin-alfa färgar in tumörer som har mutation i gener som kopplas till pseudohypoxi-klustret (med en ökad risk för framtida metastasering jämfört med tumörer som uppvisar mutationer i kinas-relaterade gener) [9, 10].
Feokromocytom kan uppkomma i komposita varianter som blandformer med andra ovanliga primärtumörer i binjuren, såsom ganglioneurom och neuroblastom. Dessa komposita varianter ger sig oftast tillkänna genom sin distinkta histomorfologi, och dessa tillstånd listas i en separat del av dokumentet nedan.
Ett heltäckande mikroskopiutlåtande om feokromocytom och abdominala paragangliom ska innefatta följande parametrar:
- Förekomst av multicentricitet.
- En generell histologisk beskrivning av lesionens eller lesionernas utseende (växtmönster, pleomorfism, hyperkromasi, ökad cellularitet, monomorfi, mitostal, atypiska mitoser, äkta tumörnekros, småcellighet och fokal spolcellighet.)
- Förekomst av komposit tumörform (ganglioneurom, ganglioneuroblastom, neuroblastom eller perifer nervskidetumör).
- Förekomst av tumörkapsel.
- Förekomst av kapselinfiltration.
- Förekomst av blodkärlsinvasion inom kapselplan.
- Förekomst av extraadrenal extension (oftast fettväv).
- Förekomst av tumörväxt i resektionsytor.
- Relevanta patologiska förändringar i icke-tumoröst parenkym (adrenomedullär hyperplasi, patologiska barknoduli, inflammation etc.)
- Vid medföljande lymfkörtlar: antal och status.
- I fall med lymfkörtelmetastaser: den största metastasens storlek samt förekomst av periglandulär växt.
Tabell 21. Pheochromocytoma of the Adrenal Gland Scaled Score 4 [18]
|
Stora nästen/kompakt växtmönster |
2 p |
|
Tumörnekros |
2 p |
|
Hög cellularitet |
2 p |
|
Cellulär monotoni |
2 p |
|
Spolcellighet (även fokal) |
2 p |
|
> 3 mitoser/10 high power fields |
2 p |
|
Atypiska mitoser |
2 p |
|
Periadrenal fettvävsinfiltration |
2 p |
|
Kärlinvasion |
1 p |
|
Kapselinvasion |
1 p |
|
Nukleär pleomorfism |
1 p |
|
Nukleär hyperkromasi |
1 p |
Totalt 20 poäng. Färre än 4 poäng indikerar liten risk för framtida metastasering. Algoritmen har bristande reproducerbarhet.
Andra primärdiagnoser att beakta
Myelolipom
En välavgränsad, benign tumör uppbyggd av fettväv och benmärgskomponent. Förkalkning och osseös metaplasi kan ses. Näst vanligaste tumören i binjurebarken. Kan förekomma i samband med binjurebarksadenom.
Schwannom
Benign tumör med nervskidedifferentiering. Ovanlig, cirka 60 fall rapporterade i vetenskaplig litteratur. Uppvisar klassisk histologi med spolceller arrangerade i Antoni A-mönster (cellulära) och Antoni B-mönster (löst organiserade). Positiv S100- och SOX10-färgning indikerar diagnosen.
Perifera neuroblastiska tumörer
En grupp tumörer som utgår från neuralrörets celler längs med sympatiska gränssträngen och binjuremärgen. Innefattar ganglioneurom, ganglioneuroblastom och neuroblastom. Histologiskt ses vid ganglioneurom en blandning av mogna ganglieceller och schwannceller, medan ganglioneuroblastom uppvisar neuroblastiska celler blandat med ganglioneuromal vävnad. Neuroblastom är framför allt en pediatrisk diagnos, och ses uppbyggd av neuroblastiska celler utan schwannomlikt stroma. Kan vara odifferentierade, lågt differentierade och differentierade beroende på graden av neuropil.
Gonadal stromal tumör (innefattandes granulosacellstumör och Leydigcell-tumör)
Mycket ovanliga tumörformer, det finns enbart sex fallrapporter i vetenskaplig litteratur, som alla rör postmenopausala kvinnor. Histologiskt identiska med korresponderande diagnos i ovarium. Immunhistokemiskt ses positivitet för binjurebarkmarkörer såsom SF1, melan-A, kalretinin och inhibin-alfa.
Adenomatoid tumör
Benign mesotelial tumör som förmodligen uppkommer ur mesoteliala rester. Histologiskt ses adenoida kännetecken i form av tubulära strukturer med varierande storlek och form som invändigt är beklädda med epiteloida till tillplattade endotelcellslika celler. Tumören är immunreaktiv mot mesotelcellsmarkörer men inte mot barkmarkörer
Koder och beteckningar
SNOMED-koder
|
Adrenokortikalt adenom |
M83700 |
|
Adrenokortikalt karcinom |
M83703 |
|
Adrenokortikal hyperplasi |
M72030 |
|
Gonadal stromal tumör |
M85901 |
|
Myelolipom |
M88700 |
|
Schwannom |
M95600 |
|
Neuroblastom |
M95003 |
|
Ganglioneuroblastom |
M94903 |
|
Ganglioneurom |
M94900 |
|
Feokromocytom |
M87003 |
|
Paragangliom |
M86803 |
Kvalitetsarbete för patologin
Kompetenskrav
För att säkerställa diagnostisk kvalitet bör binjuretumörer handläggas av specialister med särskild kompetens och erfarenhet av denna tumörgrupp. Läkare under utbildning kan handlägga dessa fall efter delegering av medicinskt ansvarig patolog.
Referenser
1. Mete O, Asa SL, Gill AJ, Kimura N, de Krijger RR, Tischler A. Overview of the 2022 WHO Classification of Paragangliomas and Pheochromocytomas. Endocr Pathol. 2022;33(1):90-114.
2. Mete O, Erickson LA, Juhlin CC, de Krijger RR, Sasano H, Volante M, Papotti MG. Overview of the 2022 WHO Classification of Adrenal Cortical Tumors. Endocr Pathol. 2022;33(1):155-96.
3. Amin MB, Greene FL, Edge SB, Compton CC, Gershenwald JE, Brookland RK, et al. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more "personalized" approach to cancer staging. CA: a cancer journal for clinicians. 2017;67(2):93-9.
4. Mete O, Asa SL, Giordano TJ, Papotti M, Sasano H, Volante M. Immunohistochemical Biomarkers of Adrenal Cortical Neoplasms. Endocr Pathol. 2018;29(2):137-49.
5. Juhlin CC, Bertherat J, Giordano TJ, Hammer GD, Sasano H, Mete O. What Did We Learn from the Molecular Biology of Adrenal Cortical Neoplasia? From Histopathology to Translational Genomics. Endocr Pathol. 2021;32(1):102-33.
6. Juhlin CC. Challenges in Paragangliomas and Pheochromocytomas: from Histology to Molecular Immunohistochemistry. Endocr Pathol. 2021;32(2):228-44.
7. Papathomas TG, Oudijk L, Persu A, Gill AJ, van Nederveen F, Tischler AS, et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: a multicenter interobserver variation analysis using virtual microscopy: a Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod Pathol. 2015;28(6):807-21.
8. Gill AJ, Benn DE, Chou A, Clarkson A, Muljono A, Meyer-Rochow GY, et al. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol. 2010;41(6):805-14.
9. Favier J, Meatchi T, Robidel E, Badoual C, Sibony M, Nguyen AT, et al. Carbonic anhydrase 9 immunohistochemistry as a tool to predict or validate germline and somatic VHL mutations in pheochromocytoma and paraganglioma-a retrospective and prospective study. Mod Pathol. 2020;33(1):57-64.
10. Mete O, Pakbaz S, Lerario AM, Giordano TJ, Asa SL. Significance of Alpha-inhibin Expression in Pheochromocytomas and Paragangliomas. The American journal of surgical pathology. 2021;45(9):1264-73.
11. Juhlin CC, Zedenius J, Höög A. Metastatic Neuroendocrine Neoplasms of Unknown Primary: Clues from Pathology Workup. Cancers. 2022;14(9).
12. Pennanen M, Heiskanen I, Sane T, Remes S, Mustonen H, Haglund C, Arola J. Helsinki score-a novel model for prediction of metastases in adrenocortical carcinomas. Hum Pathol. 2015;46(3):404-10.
13. Duregon E, Fassina A, Volante M, Nesi G, Santi R, Gatti G, et al. The reticulin algorithm for adrenocortical tumor diagnosis: a multicentric validation study on 245 unpublished cases. The American journal of surgical pathology. 2013;37(9):1433-40.
14. Weiss LM. Comparative histologic study of 43 metastasizing and nonmetastasizing adrenocortical tumors. The American journal of surgical pathology. 1984;8(3):163-9.
15. Lin BT, Bonsib SM, Mierau GW, Weiss LM, Medeiros LJ. Oncocytic adrenocortical neoplasms: a report of seven cases and review of the literature. The American journal of surgical pathology. 1998;22(5):603-14.
16. Wieneke JA, Thompson LD, Heffess CS. Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. The American journal of surgical pathology. 2003;27(7):867-81.
17. Williams TA, Gomez-Sanchez CE, Rainey WE, Giordano TJ, Lam AK, Marker A, et al. International Histopathology Consensus for Unilateral Primary Aldosteronism. J Clin Endocrinol Metab. 2021;106(1):42-54.
18. Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. The American journal of surgical pathology. 2002;26(5):551-66.
19. Wachtel H, Hutchens T, Baraban E, Schwartz LE, Montone K, Baloch Z, et al. Predicting Metastatic Potential in Pheochromocytoma and Paraganglioma: A Comparison of PASS and GAPP Scoring Systems. J Clin Endocrinol Metab. 2020;105(12):e4661-70.
20. Stenman A, Zedenius J, Juhlin CC. Over-diagnosis of potential malignant behavior in MEN 2A-associated pheochromocytomas using the PASS and GAPP algorithms. Langenbecks Arch Surg. 2018;403(6):785-90.
21. Kimura N, Takayanagi R, Takizawa N, Itagaki E, Katabami T, Kakoi N, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocrine-related cancer. 2014;21(3):405-14.
22. Stenman A, Zedenius J, Juhlin CC. The Value of Histological Algorithms to Predict the Malignancy Potential of Pheochromocytomas and Abdominal Paragangliomas-A Meta-Analysis and Systematic Review of the Literature. Cancers. 2019;11(2).